Medizingeräte bei Schlafapnoe
Bei Beatmung Tod
Die Geräte sollen Menschen mit Atemstillständen während des Schlafs helfen. Sie erhöhen bei manchen PatientInnen das Sterberisiko.

Medizinprüfer über Implantate
„Industrie verhindert Patientennutzen“
In Europa kommen weiter Medizinprodukte auf den Markt, die anderswo wegen ihres Risikos abgelehnt werden, rügt Deutschlands oberster Medizinprüfer Jürgen Windeler.

Skandal um defekte Brustimplantate
Sammelklage in Argentinien
300 betroffene Argentinierinnen fordern Schadenersatz wegen defekter Brustimplantate. Angeklagt sind auch der TÜV-Rheinland und die Allianz-Versicherung.
 Alle Artikel zum Thema
Alle Artikel zum Thema


Bitte registrieren Sie sich und halten Sie sich an unsere Netiquette.
Haben Sie Probleme beim Kommentieren oder Registrieren?
Dann mailen Sie uns bitte an kommune@taz.de.
Guy
Gast
Frau Eikermann trägt ohne Not zur Patientenverunsicherung bei und bewirbt die kommerziellen Dienstleistungen ihrer Kollegen.
anke
Gast
Leider ist das ja immer eine recht schwierige Sache mit solchen ethischen Grundsätzen. Wer "zuallererst nicht schaden" will, der müsste zunächst erst einmal wissen, was im konkreten Fall wohl schaden wird, vor allem aber: wann.
Der Haken an der Individualität ist leider, dass sie die Vorhersehbarkeit der Dinge erkennbar reduziert. Was allerdings immer nur dann auffällt, wenn die abgeschlossene Wette verloren wird. In sofern können Leute, die sich vorsorglich verstümmeln lassen, nur gewinnen. Dass sie nämlich ohne die OP nicht doch länger und/oder glücklicher ge- bzw. überlebt hätten, ist schlicht nicht zu beweisen. Vielleicht ist das der Grund, dass es deutlich mehr Feigheit gibt auf dieser Welt als Optimismus. Wenn Optimisten sich irren, heißt es immer: "Siehst du, hättest du mal...!" Feigheit "rechnet" sich einfach besser in Gesellschaft.
Ich denke, Nietzsche hat sich gleichzeitig geirrt und recht gehabt mit seiner Behauptung, Gott sei tot. Ein Teil der Menschheit nämlich hat Gott nicht selber und für sich persönlich abgeschafft, sondern nur einem (vermeintlichen) Zwang nachgegeben. Dieser Teil konnte im Anschluss das Loch, das durch das verfrühte Ableben ihres "Vaters" entstanden ist, einfach nicht ertragen. Sie hat kurzerhand neue Autoritäten auf die alten Throne gesetzt. Die Schulmedizin etwa, bestimmte Hollywood-Schauspieler oder gleich sich selbst. "Was er/sie/es/glaubt, was ich glaube, ist wahr", wird seither behauptet. So kommt es, dass wahlweise eine 80%-ige oder eine 5%-ige Wahrscheinlichkeit als 100%-iger Grund dafür taugt, die seltsamsten Dinge zu tun oder zu lassen. Wer glaubt, wird entweder (kurzzeitig) selig, oder (ebenso kurzzeitig) zum Zweifler. Die Phasen des Zweifels vergisst man offenbar leichter.
Dass Nietzsche diese Begleiterscheinungen seines Postulats nicht umfassend thematisiert hat, könnte man ihm natürlich vorwerfen – wenn man Gott wäre. Und entsprechend unfehlbar.
Empfehlungen zu § 218-Reform
„Erwarte, dass sie diese umsetzen“
Die Juristin Liane Wörner gehörte zur Expert_innengruppe für die Reform des Abtreibungsrechts. Im Gespräch weist sie die Kritik an der Kommission ab.
Umfrage zu Abtreibungen in Deutschland
Große Mehrheit für Legalisierung
Zentralratspräsident über Nahost-Krise
„Ein Gewitter reinigt die Luft“
Reform des Klimaschutzgesetzes
Ran ans Auto!
Reform des Klimaschutzgesetzes
Verwässertes Klimagesetz kommt
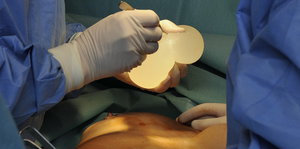
Debatte Brustimplantate: Zuallererst nicht schaden
Am Dienstag soll in Marseille der Brustimplantante-Prozess enden. Noch immer fehlen EU-weit unabhängige Tests für solche Medizinprodukte.
Auf der Brust kein Problem, drin aber eventuell schon: Fremdkörper Bild: ap
Franz P. ist 75 Jahre alt. Vor einigen Jahren benötigte er ein künstliches Hüftgelenk (Endoprothese). Sein Arzt erklärte ihm damals, dass dies ein häufiger Eingriff sei und dass er sich nicht sorgen müsse. Er war trotzdem besorgt, hatte Angst vor Komplikationen. Er informierte sich über die Narkose, die Wahrscheinlichkeit für Bluttransfusionen, die Versorgung nach der Operation, die Lebensdauer der Prothese.
Über die Prothese selbst machte er sich keine Gedanken. Es erschien ihm selbstverständlich, dass Materialien, die über einen so langen Zeitraum im Körper eines Menschen verbleiben, mindestens so gut getestet wurden wie die Tabletten, die er wegen seiner Hüftschmerzen so häufig nehmen musste. Jetzt ist er beunruhigt. Immer wieder hat er von fehlerhaften Medizinprodukten gelesen, die Schaden im menschlichen Körper anrichten können.
Giftige Schwermetalle aus Metall-Endoprothesen, Brustimplantate, die mit Industriesilikon gefüllt wurden, Versagen von Messsystemen für Diabetiker: Die Liste der Berichte über fehlerhafte Medizinprodukte ist lang. Sie erstrecken sich über zahlreiche medizinische Bereiche und betreffen unterschiedlichste Produkte.
Allen gemeinsam ist, dass sie ein deutliches Signal dafür sind, dass das europäische Regulationssystem für Medizinprodukte in seiner aktuellen Form nicht ausreichend ist, eine sichere und effektive Patientenversorgung zu gewährleisten. Seit Jahren schon wird dies von Experten moniert. Zentrale Kritikpunkte waren immer wieder das uneinheitliche, interessengesteuerte und intransparente Zertifizierungsverfahren, die oft mangelhafte Evidenz sowie fehlende Langzeitbeobachtungen der Patienten.
Die Europäische Kommission als zuständiges Organ hat darauf reagiert und im September 2012 einen Verordnungsentwurf veröffentlicht. Dieser nimmt Forderungen etwa nach einer besseren Langzeitbeobachtung auf, lässt aber andere Punkte unangetastet oder lässt es in der Umsetzung an Konsequenz vermissen.
Petition von Experten
Daher hat sich eine Gruppe klinischer und methodischer Experten aus ganz Europa zusammengeschlossen und gemeinsam eine Petition verfasst, die drei zentrale Forderungen an die Brüsseler Kommission enthält:
1. Zentralisierung des Regulationsprozesses und Unabhängigkeit der Bewertung: Aktuell erhalten Medizinprodukte ein Prüfsiegel (CE-Kennzeichnung) von einer sogenannten benannten Stelle. Danach können diese Medizinprodukte im europäischen Markt vertrieben werden. Zurzeit gibt es über 80 solcher benannten Stellen, für die es keine einheitlichen Prüfkriterien gibt. Die Hersteller können selbst wählen, bei welcher Stelle sie ihr Präparat prüfen lassen möchten.
Ein solches System, in dem die benannten Stellen miteinander im wirtschaftlichen Wettbewerb stehen und in denen die Prüfung von den Herstellern beauftragt und bezahlt wird, bietet natürlich eine Chance, besonders niederschwellige Prüfungen anzubieten beziehungsweise solche auszuwählen.
MICHAELA EIKERMANN
leitet die Sektion Evidenzbasierte Medizin der Uni Witten/Herdecke. Die Petition an die EU ist hier zu finden.
Vor Kurzem wurde vom British Medical Journal und dem Daily Telegraph auf erschreckende Weise vorgeführt, wie korrupt dieses System sein kann: Die Journalisten gaben sich als Hersteller einer fiktiven Hüftprothese aus. Sie legten die Daten eines Produkts vor, das bereits zwei Jahre zuvor wegen Fehlerhaftigkeit vom Markt genommen worden war. Trotzdem hätten sie von einer benannten Stelle eine CE-Kennzeichnung erhalten und ein fehlerhaftes Produkt auf dem europäischen Markt vertreiben können.
Daher ist es aus Sicht der Experten essenziell, dass der Regulationsprozess standardisiert und unabhängig erfolgt. Dies lässt sich am besten durch einen zentralisierten europäischen Prozess erreichen. Im aktuellen Verordnungsentwurf wird jedoch kein solcher unabhängiger zentraler Regulationsprozess vorgeschrieben, sondern am System der benannten Stellen festgehalten. Zwar sollen diese stärker durch Aufsichtsbehörden kontrolliert werden, es soll jedoch auch für Hochrisikoprodukte kein Verfahren eingeführt werden, das mit dem der Arzneimittelregulation vergleichbar wäre.
Forderung nach klinischen Studien
2. Stärkere Verpflichtung zur Einbeziehung von hochwertiger Evidenz zum patientenrelevanten Nutzen: Um eine CE-Kennzeichnung zu erhalten und damit ein Medizinprodukt auf dem europäischen Markt zu vertreiben, ist es aktuell selbst für Hochrisikoprodukte nicht zwingend notwendig, dass Hersteller methodisch hochwertige klinische Studien an ausreichend großen Patientengruppen vorlegen. Es wird nach wie vor nicht verbindlich gefordert, dass Medizinprodukte nur zugelassen werden können, wenn valide Daten zum Nutzen und Schaden vorliegen. Auch hier besteht ein nicht nachvollziehbarer Unterschied zur Arzneimittelregulation.
Transparenz ist nötig
3. Transparenz des Bewertungsprozesses und der Ergebnisse: Aktuell und auch nach der geplanten Änderung sind weder die klinischen Daten zu einem Medizinprodukt noch die Ergebnisse der Bewertungen im Rahmen der Regulationsprozesses öffentlich zugänglich. Es kann damit nicht nachvollzogen werden, auf welcher Datenbasis ein Medizinprodukt zugelassen wurde. Dies ist umso kritischer zu sehen, da keine verbindliche Vorgabe bezüglich einer hochwertigen Evidenzbasis existiert.
Patienten und Ärzte müssen sich darauf verlassen können, dass ein zugelassenes Medizinprodukt ausreichend geprüft ist. Natürlich kann man argumentieren, dass es nur schwarze Schafe sind, die den Ruf eines eigentlich gut funktionierenden Systems ruinieren. Man kann auch argumentieren, dass die Einführung eines zentralen Zulassungsverfahrens viel zu teuer ist. Und man kann auch prophezeihen, dass ein verschärfter Regulationsprozess länger dauert und innovationsfeindlich ist.
Sicher ist jedoch, dass das fast schon etwas verbraucht klingende „primum nil nocere“ (zuallererst nicht schaden) immer noch der ethische Grundsatz ärztlichen Handelns sein muss. Sicherstellen kann dies der Arzt offensichtlich nicht allein, dazu muss der Grundsatz auch von denjenigen angenommen werden, die Medizinprodukte herstellen oder für die Anwendung am Patienten freigeben.
Fehler auf taz.de entdeckt?
Wir freuen uns über eine Mail an fehlerhinweis@taz.de!
Inhaltliches Feedback?
Gerne als Leser*innenkommentar unter dem Text auf taz.de oder über das Kontaktformular.
Kommentar von
Michaela Eikermann
Themen